Identificación de Metabolitos Secundarios Liquénicos por Cromatografía en Capa Fina (CCF)
Frank Bungartz1,2,3, Alba Yánez-Ayabaca3,4,5 y María de los Ángeles Herrera-Campos6
1Biodiversity Integration Knowledge Center, Arizona State University, PO Box 874108, Arizona State University, Tempe, AZ 85287-4108, USA
2Charles Darwin Foundation for the Galapagos Islands, Puerto Ayora, Ecuador
3Instituto Nacional de Biodiversidad (INABIO), Quito, Ecuador
frank.bungartz@asu.edu
4Universidad Central del Ecuador, Ciudadela Universitaria, Jerónimo Leyton s/n, Quito, Ecuador
5Herbario Nacional del Ecuador, Quito, Ecuador
albayanez8@gmail.com
6Departamento de Botánica, Instituto de Biología, Universidad Nacional Autónoma de México. Apartado Postal 70-367, 04510 México, Cd. de México, México
mahc@ib.unam.mx
Table Of Contents
_______________________________________________________________________________________
Introducción
Las sustancias liquénicas son productos orgánicos del metabolismo primario y del metabolismo secundario. Los primeros son producidos por el fotobionte y el micobionte y se depositan intracelularmente, mientras que los segundos son productos fúngicos, que se depositan en las paredes celulares e incluyen para y meta-depsidas, depsidonas, bencil ésteres, dibenzofuranos, ácidos úsnicos, xantonas, antraquinonas, terpenoides y ácido pulvínico y sus derivados y ácidos alifáticos y son sintetizados a través de tres vías metabólicas secundarias: vía acetato-malonato, vía del ácido siquímico y vía del ácido mevalónico. Se conocen alrededor de 700 metabolitos secundarios liquénicos, de los cuales entre 50 y 60 también han sido encontrados en hongos no liquenizados y /o en plantas vasculares.
La cromatografía en capa fina CCF (TLC por sus siglas en inglés) es una técnica relativamente simple, no muy costosa y probablemente la más utilizada para la identificación de las sustancias liquénicas, sin embargo no es suficientemente sensible para detectar aquéllas presentes en bajas concentraciones que frecuentemente no son evidentes en la placa cromatográfica.
El método consiste, de manera general, en correr el extracto acetónico de las sustancias liquénicas en una placa cromatográfica con un solvente que se desplaza por capilaridad a lo largo de la misma. Las sustancias se separan en la placa de acuerdo a su afinidad por el solvente y se evidencian como manchas de diferentes características bajo luz visible y luz ultravioleta de onda corta y larga antes y después de un proceso de revelado en ácido sulfúrico a alta temperatura en un horno de laboratorio
Las características de estas manchas que deben ser registradas para identificar las sustancias liquénicas son la distancia que recorre cada una en la placa a partir del origen o línea base y su color al observarlas bajo luz visible y UV.
La distancia recorrida por las manchas puede ser expresada como el valor Rf (valor de retención), cuyo valor absoluto puede variar debido a las condiciones particulares de cada laboratorio. Para evitar estas variaciones, se dividen las placas cromatográficas por valores Rf, tomando como referencia sustancias controles valores Rf conocidos como el ácido norestíctico y la atranorina (Fig. 1). Otra posibilidad es no usar los valores absolutos, sino calcular los valores Rf relativos con la siguiente fórmula

El color de las manchas en luz visible y ultravioleta de onda larga (366 nm) y onda corta (254 nm) es comúnmente diferente bajo cada una. Muchas sustancias tienen fluorescencia característica bajo la luz UV que incluso puede ser observada aún antes del proceso de revelado con el ácido sulfúrico. De hecho, casi todas las sustancias son visibles como manchas obscuras al exponer la placa a luz UV de onda corta, pero al observarla bajo UV de onda larga sólo algunas fluorescen de manera característica, lo cual puede cambiar después del tratamiento con él ácido sulfúrico.
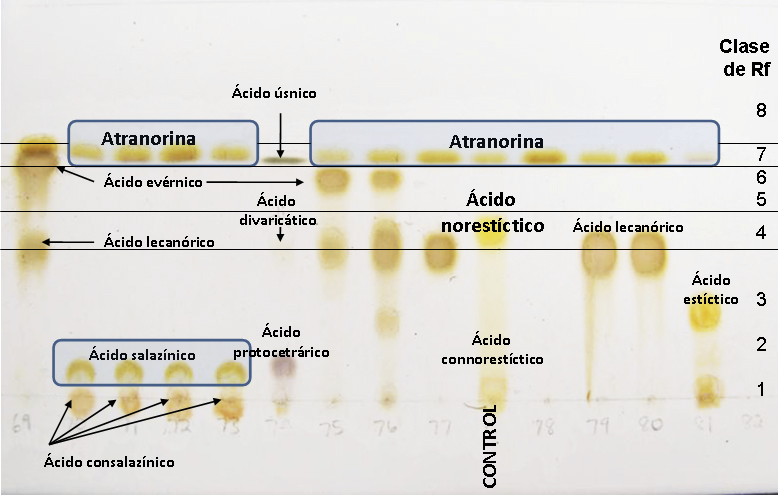
Figura 1. División de la placa por clases Rf en función de atranorina y ácido norestíctico como controles Las sustancias con un Rf igual al ácido norestíctico pertenecen a la clase de Rf 4 y las sustancias con un Rf parecido a atranorina a la clase 7. Las sustancias con Rf de 0 (básicamente no saliendo del origen de la placa) pertenecen a la clase 1. Según estos controles la región entre 1 y 4 se divide en las clases 2 y 3 y el área entre 4 y 7 contiene las clases 5 y 6. Cualquier sustancia que suba por arriba de Rf7 pertenece a la clase Rf 8.
_______________________________________________________________________________________
Procedimiento
(1) Uso de controles y/o ejemplares de química conocida para comparar
Se recomienda usar la atranorina y el ácido norestíctico como controles generales, aunque se pueden agregar otros para ayudar a la identificación de las manchas. Estos controles pueden ser extractos específicos purificados industrialmente o muestras de líquenes cuya química secundaria es conocida.
Generalmente, para verificar si una sustancia específica está presente en una muestra, es útil correr una muestra cuya química se desconoce al lado de una muestra con química conocida para compararlas. Por ejemplo, en muchas regiones tropicales Pseudocyphellaria aurata es un liquen muy común. El amarillo de su médula corresponde al ácido pulvínico. Para verificar que éste también está presente en otro liquen con pigmento amarillo intenso se puede correr directamente al lado de Pseudocyphellaria aurata. A veces este procedimiento de comparar muestras conocidas con muestras desconocidas es más fácil, más rápido y más confiable que analizar las placas solamente según sus valores o clases de Rf.
_______________________________________________________________________________________
(2) Preparación de muestras
Al estereoscopio, se toma una pequeña porción del liquen, seca y libre de sustrato y se coloca en un vial numerado, ¡asegurándose no colocar una mezcla de diferentes especies!
A temperatura ambiente, se añade a cada vial una gota de acetona o lo estrictamente necesario para cubrir la muestra. Para muestras muy absorbentes o de extractos muy pálidos se requerirán gotas adicionales que se agregan conforme las anteriores se evaporen. Algunas sustancias secundarias de los líquenes disuelven muy rápido en acetona y generalmente después de dos o tres minutos de añadir la acetona, los extractos están listos para ser analizados. Sin embargo algunas sustancias secundarias son poco solubles y en este caso poner los tubos de extracción con la muestra y el acetona en un baño de agua hirviendo por algunos segundos puede significativamente mejorar la extracción aunque es muy importante que no se aplica el calor por mucho tiempo.
_______________________________________________________________________________________
(3) Preparación de los solventes
Se utilizan distintas mezclas de solventes -en proporciones indicadas a continuación entre ( )-y deben ser preparadas en pequeñas cantidades dentro de una campana de extracción de vapores utilizando cristalería completamente limpia y seca.
Solvente A: tolueno/dioxano/ácido acético (180:45:5):
El dioxano es una sustancia higroscópica que con el tiempo absorbe el agua, provocando que el solvente se deteriore dentro de unas semanas, por lo que se recomienda utilizarlo rápidamente. En regiones tropicales con alta humedad ambiental es recomendable usarlo fresco.
Solvente B’: hexano/metil-tert-butil éter/ácido fórmico (140:72:18):
En el procedimiento original se usa dietil éter en vez de metil-tert-butil éter. El dietil éter es una sustancia muy peligrosa, por lo que ha sido reemplazada con el solvente B’. El solvente B se echa a perder en menos de 6 horas, mientras que el B’ puede ser utilizado durante cuatro o cinco días. Por seguridad también se puede reemplazar el hexano por ciclohexano.
Solvente C: tolueno/ácido acético (170:30; o 200:30):
El solvente estándar para CCF de rutina; es ampliamente utilizado porque permite distinguir la mayoría de las sustancias químicas y es estable durante varias semanas.
Solventes especiales:
Los solventes E y G son usados para diferenciar sustancias que no distinguibles con el uso de los solventes anteriores.
Solvente E: ciclohexano/acetato de etilo (75:25)
El solvente E discrimina varios derivados no polares de compuestos liquénicos y sustancias cuyos valores Rf son muy altos en los solventes A, B, B’ y C. Es necesario prepararlo diariamente.
Solvente G: tolueno/acetato de etilo/ácido fórmico (139:83:8)
El solvente G discrimina con mejor resolución metabolitos que salen con Rf bajo en otros solventes.
Solvente F: acetato de etilo / cyclohexano 1: 1 (Elix & Crook 1992).
El solvente F se usa para analizar xantones, además ayuda para distinguir atranorina de chloroatranorina.
En caso de utilizar los solventes A y B, colocar un papel filtro en la cara posterior del tanque para ayudar a uniformizar su distribución. Al introducir la placa en el tanque la superficie de sílica debe quedar de frente al papel.
Raramente, se usan también otros solventes para una diferenciación mejor de sustancias particulares como xantonas o terpenos lo cuales son generalmente más difícil de analizar (Orange et al. 2010).
_______________________________________________________________________________________
(4) Preparación del tanque
Algunos autores recomiendan antes de utilizar los solventes B y C, prevenir el desarrollo de frentes secundarios colocando las placas con las muestras en un tanque con ácido fórmico por cinco minutos (para el solvente B) y durante 10 minutos con ácido acético glacial (para el C). Se debe evitar que dichos ácidos toquen la placa, por lo que han de usarse cuentas o viales de vidrio en el fondo del tanque para soportarla y de preferencia colocarla de manera invertida, con la línea basal hacia arriba.
Una vez preparada la mezcla a utilizar, en un tanque para cromatografía se vacía una pequeña cantidad, que permita alcanzar 1 cm de profundidad. Se engrasa el borde del tanque con grasa silicónica para sellarlo herméticamente y el solvente se deja reposar dos horas antes de usarlo.
_______________________________________________________________________________________
(5) Preparación de las placas
Las placas cromatográficas más comúnmente usadas para un tanque estándar miden 20 x 20 cm (para revisar las placas es importante comprarlas con un indicador de fluorescencia en luz de UV corta (F254), ej. Merck silica gel 60 F254 pre-coated 20 × 20 cm). Los tanques estándar son un jarrón recto de vidrio que requiere una cantidad relativamente alta de solvente. Para ahorrar solvente es muy recomendable usar un tanque plano especial de HPTLC [High Performance Thin-layer Chromatography. Aunque el tanque es inicialmente más caro, a largo plazo representa un ahorro en la compra de solventes (tanques planos de HPTLC se puede comprar de CAMAG o de DESAGA). Las placas para HPTLC tienen un grano más fino de sílica, mejor resolución y típicamente miden solamente 10 x 10 cm.]
En caso de que se analicen pocas muestras, es posible cortar las placas por el reverso pasando firmemente y una sola vez con un cortador y utilizando una regla como guía. Cortar placas de vidrio generalmente no es fácil y alternativamente se puede usar placas con una base de aluminio, también conocidas como cromatofolios, que pueden cortarse con tijera (desventaja: en estas placas no se puede ver manchas de ácidos grasos que solamente son observables en placas con una base de vidrio).
A una distancia de 2cm del borde inferior de la placa, dibujar con lápiz SUAVE la línea base de lado a lado, marcando ligeramente a cada centímetro la posición donde se colocará cada muestra de los extractos. Si se desea, también se puede trazar otra línea en la parte superior de la placa para indicar hasta dónde llegará el frente del solvente (típicamente a 10-15 cm de la línea base; la resolución de las manchas en la placa es generalmente mejor con la distancia, pero a mayor distancia los artefactos, como efectos del borde de la placa, son también más pronunciados).
Con un tubo capilar, tomar una muestra del extracto acetónico y cuidadosamente colocar una pequeña gota en el lugar correspondiente en la placa. No usar el mismo capilar para distintas muestras.
Repetir el procedimiento hasta que una mancha de sustancia liquénica pueda distinguirse en la placa. Se puede asegurar que hay suficiente muestra si las manchas son evidentes al ver la placa por el reverso a contraluz (Fig. 2).
Figura 2. Reverso de la placa, línea de origen vista a contraluz.
_______________________________________________________________________________________
(6) Corriendo las placas
La placa se coloca rectamente en el tanque y se deja correr el solvente, cuidando que el frente ¡no sobrepase el borde de la misma! Una vez que el frente del solvente alcanza la distancia adecuada (10-15 cm), remover la placa del tanque y dejarla secar 30 minutos.
_______________________________________________________________________________________
(7) Interpretación de las manchas
Un recurso bien útil para la analisis es el programa MYTABOLITES, disponible para bajar con instrucciones detalladas directamente desde de la pagina aquí. La base de datos incluido en el programa ayuda mucho en la interpretación de placas de cromatografía de capa fina para el análisis de metabolitos secundarios de líquenes.
Observación ácidos grasos
A continuación, la placa de vidrio seca se rocía con agua para detectar los ácidos grasos, los cuales serán visibles como manchas blancas opacas en el fondo húmedo y translúcido de la placa. Trazar un círculo alrededor de la mancha y cruzarlo. Resulta de utilidad observar la placa contra una superficie negra.
Observación en luz UV antes de hornear con H2SO4
Nuevamente permitir que la placa se seque y observarla bajo UV de onda corta y encerrar cada mancha en un círculo. Inmediatamente, observarla bajo UV de onda larga y marcar con un círculo punteado cada mancha, registrando el color, la presencia de halos y la intensidad de la fluorescencia con +, ++, +++. En el caso de manchas obscuras, colocar un – en el centro del círculo punteado (esta documentación es necesario especialmente cuando no es posible documentarlo con fotografía digital).
Tratamiento con H2SO4
Una vez registrada la información anterior, aplicar ácido sulfúrico al 10% a la placa y dejarla secar para meterla al horno a 100 o 110°C durante 8 minutos aproximadamente, cuidando que la placa no se queme y revisando el desarrollo de los colores de las manchas, los cuales deben aparecer nítidos.
Observación en luz visible y en luz UV después de hornear con H2SO4
En cuanto se revelan las manchas coloridas, sacar la placa del horno, dejarla enfriar y registrar sus colores bajo luz visible. Repetir la lectura bajo UV larga y registrar cambios de color de manchas ya detectadas y/o las características de manchas nuevas.
Algunos colores pueden cambiar rápida o lentamente, por lo que se recomienda revisar la placa repetidamente después de un tiempo y anotar los cambios.
Identificación de las sustancias secundarias
Con la información obtenida durante los diferentes pasos de la CCF es posible identificar para la gran mayoría de las sustancias: la clase Rf, el Rf relativo, su presencia o ausencia en luz UV antes de hornear, el color después en luz UV y luz visible. Asimismo, se puede añadir la información de las pruebas de tinción y fluorescencia del ejemplar de liquen para identificar las sustancias presentes.
Generalmente la interpretación de las placas para la identificación de las sustancias secundarias es el paso más difícil en el análisis y no siempre es posible identificar todas las manchas correctamente. Todavía hay varias sustancias desconocidas y el descubrimiento de nuevas continúa hasta la fecha.
En la mayoría de casos la CCF rutinaria es suficiente para confirmar la presencia o ausencia de una sustancia específica en un ejemplar. Dicho ejemplar es identificado preliminarmente y se sabe que tal especie está caracterizada por la presencia de una sustancia característica, por lo que para confirmar la identificación no se requiere más que una cromatografía en el solvente estándar C, corriendo la muestra problema al lado de la muestra con sustancias conocidas para verificar su presencia en ambas y saber si son idénticas o no. En otros casos, cuando se trata de sustancias menos conocidas, frecuentemente es necesario correr los extractos en distintos solventes.
La variación de las condiciones en el laboratorio, la extracción, la aplicación en las placas, la humedad, la edad del solvente, la pureza de los reactivos y muchos otros factores pueden causar que los valores Rf relativo calculados no coincidan con las valores estándar. Por ejemplo, el Rf relativo estándar de los controles del ácido norestíctico en C es 30 y el de la atranorina es 79. Si los valores reales de estos controles muestran una discrepancia, es necesario ajustar también todos los valores de las otras sustancias encontradas en la placa.
El cálculo y el registro de los valores Rf relativos para cada mancha pueden hacerse con el programa WinTab 64bit. Este programa incluye una base de datos enorme de valores Rf relativos de las manchas, colores y fluorescencias características antes y después del tratamiento con H2SO4. También permite calcular el Rf y ajustar (‘cune”) los valores si éstos no siguen el estándar en la base de datos del programa. El programa es útil para generar automáticamente sugerencias de posibles sustancias, pero la estandarización e interpretación de los colores es todavía problemática y limitada. La interpretación de un color generalmente depende del observador y es subjetivo, lo cual también ocurre cuando se utiliza información publicada (Elix 2018). Además, variaciones experimentales como el tiempo de horneado y la concentración de la sustancia afectan el desarrollo del color de mancha. Muchas manchas tienen una franja de diferente color que es característica, un detalle no usado en el programa.
Una vez digitalizada la placa, es teóricamente posible usar programas como Adobe Photoshop con su “color picker” para medir exactamente los valores RGB de un color y para la interpretación es muy recomendable establecer un registro digital de imágenes de las placas de CCF.
_______________________________________________________________________________________
(8) Documentación
Al paso del tiempo, los colores de las manchas en las placas palidecen gradualmente, por lo que se sugiere guardarlas en protectores de hojas y escanearlas o tomar fotografías digitales para una buena documentación. Con una cámara digital es posible preservar las imágenes de las placas en luz visible y en luz UV antes y después del tratamiento con H2SO4. Esta documentación facilita mucho la interpretación del análisis. Por ejemplo, en lugar de utilizar diferentes símbolos para marcar las manchas en las placas, se pueden colocar las diferentes fotos lado a lado y así documentar en detalle todos las diferentes reacciones: (1) directamente después del corrimiento y antes del tratamiento con H2O2 en luz UV de onda larga, (2) en luz UV de onda corta, (3) después del tratamiento con H2O2 en luz UV corta y (4) en luz visible (Fig. 3).
Es recomendable conservar los viales o tubos Eppendorf con el extracto, en caso de requerir repetir el análisis con distintos solventes.
Una vez analizadas las muestras, deberá anotarse la información de las sustancias identificadas e incorporarla al sobre con el ejemplar analizado.
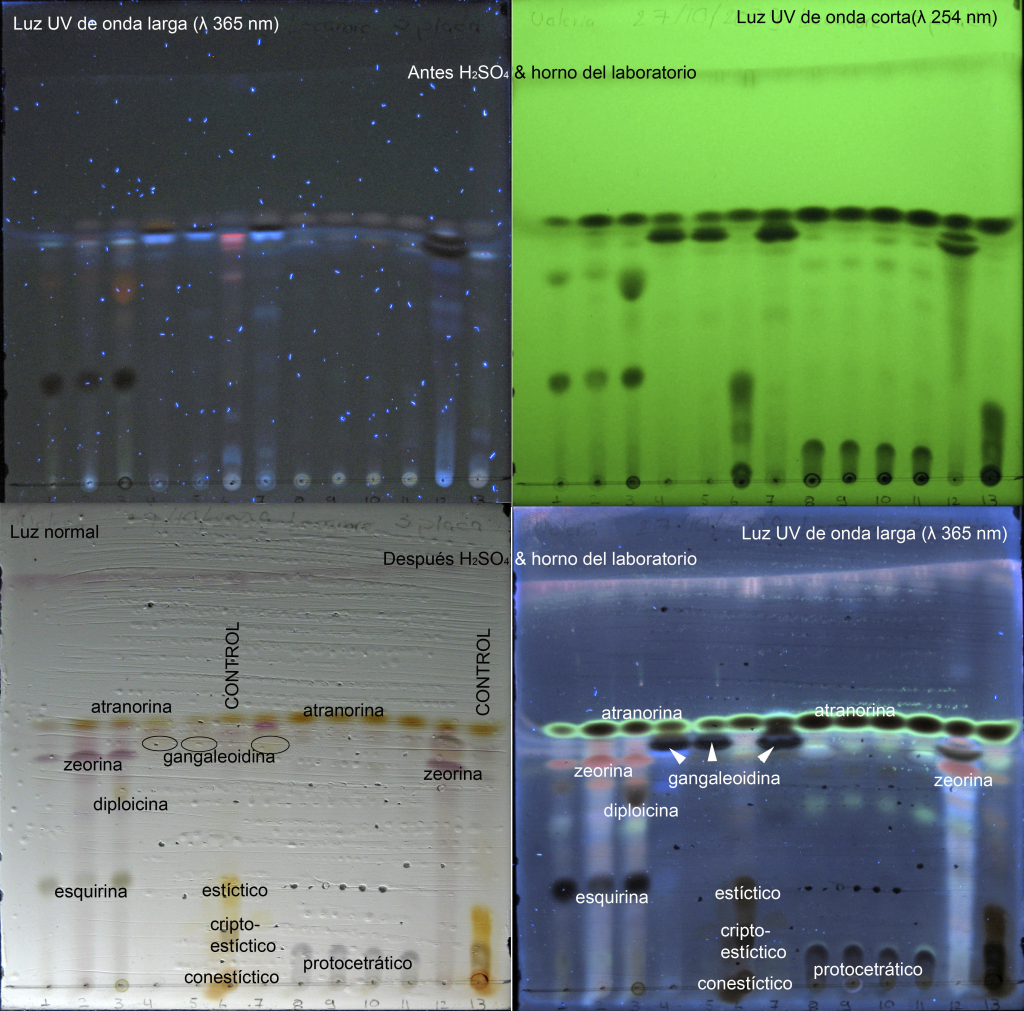
Figura 3. Comparación de una placa de HPTLC en solvente C, en luz visible con luz UV de diferentes ondas ( l 254 y 365 nm) antes y después de tratamiento con H2SO4 y quema en el horno del laboratorio.
_______________________________________________________________________________________
(9) Requerimientos y Suministros
- Campana de extracción de vapores, horno de laboratorio, estereoscopio
- Lámpara de luz ultravioleta de onda larga (λ 365 nm) y onda corta (λ 254 nm)
- Placas de vidrio y/o aluminio para cromatografía en placa fina cubiertas con sílica gel amorfo y con indicador de fluorescencia (λ 254 nm)
- Tanques de vidrio con tapa para cromatografía (o tanque plano de HPTLC)
- Papel filtro (para cara posterior del tanque, si se usa solvente A o B; no necesario para HPTLC)
- Grasa de sílica (para sellar el tanque; no necesario para HPTLC)
- Cristalería (matraces, pipetas, probetas, vasos de precipitado)
- Tubos capilares, viales o tubos Eppendorf
- Gradilla, Lápiz suave
- Reactivos: acetona (para extraer); para las solventes – acetato de etilo, ciclohexano, dioxano, hexano, metil-tert-butil-éter, tolueno. Ácidos: acético, acético glacial, fórmico y sulfúrico (10%)
- Esponja, brocha fina o rociador para aplicación de 10% H2SO4 (se necesita una campana de extracción y gafas de protección si se usa rociador), guantes
- Agua destilada en rociador, cartoncillo negro (o superficie obscura para la observación de ácidos grasos)
- Sustancias control o ejemplares de líquenes de química conocida
- Cámara digital (o escáner)
_______________________________________________________________________________________
(10) Resumen corto de procedimientos (indicando las pausas posibles)
(1) Extracción de las muestras con acetona en tubos Eppendorf (posible pausa: varios días, una vez las muestras están dentro de los tubos se puede guardarlas indefinidamente y aplicar acetona otra vez cuando el extracto se necesite).
(2) Preparar placas y aplicar extracciones (posible pausa: se puede guardar las placas en un lugar seco por unos días).
(3) Preparación del tanque y del solvente (pausa no posible: para la mayoría de los solventes es mejor prepararlos fresco y utilizarlos el mismo día).
(4) Corrimiento y lectura de las placas (inmediatamente después de la evaporación completa de las solventes en UV de onda larga (λ 365 nm) y onda corta (λ 254 nm); aplicación de H2O para visualización de ácidos grasos; documentación en luz UV con fotos; pausa no posible).
(5) Aplicación de H2SO4; secar completamente; colocar las placas en horno de laboratorio (100-110ºC, típicamente 8 minutos, pero revisar el color de las manchas; pausa no posible)
(6) Lectura de las placas después de H2SO4 y horneado: luz visible, luz UV de onda corta (λ 254 nm); toma de fotos (pausa no posible: las colores empiezan cambiar inmediatamente después y son más característicos en cuanto se saca del horno, cuando la placa está suficiente fría para tocarla; algunas manchas cambian el color característicamente en unos 20-30 minutos después, ¡anotar estos cambios!).
_______________________________________________________________________________________
Literatura y Recursos
La información más amplia y más actual sobre el análisis microquímico se encuentra en:
Orange, A, James, PW & White, FJ (2010) Microchemical methods for the identification of lichens, second edition with additions and corrections. British Lichen Society, London. Second edition with additions and corrections. Published by the British Lichen Society, available for download here.
Elix, JA (2022) Catalogue of standardized chromatographic data and biosynthetic relationships for lichen substances. Sixth Edition. Published by the author, Canberra, available for download here.
Lafferty, D., Bungartz, F., Elix, J.A. & Schumm, F. (2024) Mytabolites – a program developed at Arizona State University for the interpretation thin-layer chromatography plates, for the analysis of secondary metabolites from lichens, based on an original concept published by E. Mietzsch, H.T. Lumbsch & J.A. Elix. Help & Resources for the Consortium of Lichen Herbaria, available at https://help.lichenportal.org/index.php/en/resources/metabolites/
El programa MYTABOLITES se puede bajar aquí.
_______________________________________________________________________________________
Otras Referencias
Nylander W (1867) Hypochlorite of lime and hydrate of potash, two new criteria in the study of lichens. Linnean Society’s Journal of Botany 9: 357-365.
White FJ, James PW (1985) New Guide to microchemical techniques for the identification of lichen substances. Bulletin British Lichen Society 57: 41.
Culberson CF (1972) Improved conditions and new data for the identification of lichen products by a standardized thin-layer chromatographic method. Journal of Chromatography 72: 113-125.
Culberson CF, Kristinsson HD (1970) A standardized method for the identification of lichen products. Journal of Chromatography 46: 85-93.
Culberson CF, Culberson WL, Johnson A (1981) A standardized TLC analysis of β-orcinol depsidones. Bryologist 84: 16-29.
Culberson, CF, Ammann, K (1979) Standardmethode zur Dünnschichtchromatographie von Flechtensubstanzen. Herzogia 5: 1-24.
Culberson CF, Johnson A (1982) Substitution of methyl tert-butyl ether for diethyl ether in the standardized thin-layer chromatic method for lichen products. Journal of Chromatography 238: 483-487.
Elix, JA (2022) Catalogue of standardized chromatographic data and biosynthetic relationships for lichen substances. Sixth Edition. Published by the author, Canberra, available for download here.
Huneck S, Yoshimura I (1996) Identification of Lichen Substances. Springer, Heidelberg.
Orange, A, James, PW & White, FJ (2001) Microchemical methods for the identification of lichens. British Lichen Society, London.
Orange, A, James, PW & White, FJ (2010) Microchemical methods for the identification of lichens, second edition with additions and corrections. British Lichen Society, London.
Culberson CF (1969) Chemical and botanical guide to lichen products. Chapel Hill. University of North Carolina Press.
Culberson CF (1976) Supplement to chemical and botanical guide to lichen products. Bryologist 73: 177-377.
Culberson, CF, Culberson, WL, Johnson A (1977) Second supplement to chemical and botanical guide to lichen products. Missouri Botanical Garden. St. Louis.
Mietzsch E, Lumbsch HT, Elix JE (1994) WINTABOLITES (Mactabolites for Windows). Users manual and computer program, 2nd ed. Universität Essen.
Arup U, Ekman S, Lindblom L, Mattsson JE (1993) High performance thin layer chromatography (HPTLC), an improved technique for screening lichen substances. Lichenologist 25(1): 61-71.
